! pip install rdkit rdchiral
! mkdir data/
! curl -L https://www.dropbox.com/sh/6ideflxcakrak10/AADN-TNZnuGjvwZYiLk7zvwra/schneider50k -o data/uspto50k.zip
! unzip data/uspto50k.zip -d data/
! wget https://raw.githubusercontent.com/schwallergroup/ai4chem_course/main/notebooks/08%20-%20Retrosynthesis/utils.py15 Week 8: Retrosynthetic analysis
![]()
So far we in the course we have learnt how Machine Learning can help us classify and predict properties of molecules, how to generate new molecules with desired properties, and finally how to predict the reactivity of a reaction mixture.
Central to the chemists’ work is, however, how to make molecules, and this is what we will explore in this notebook. To do retrosynthetic analysis, we start from the target molecule and try to decide what transformation would lead to the desired product.
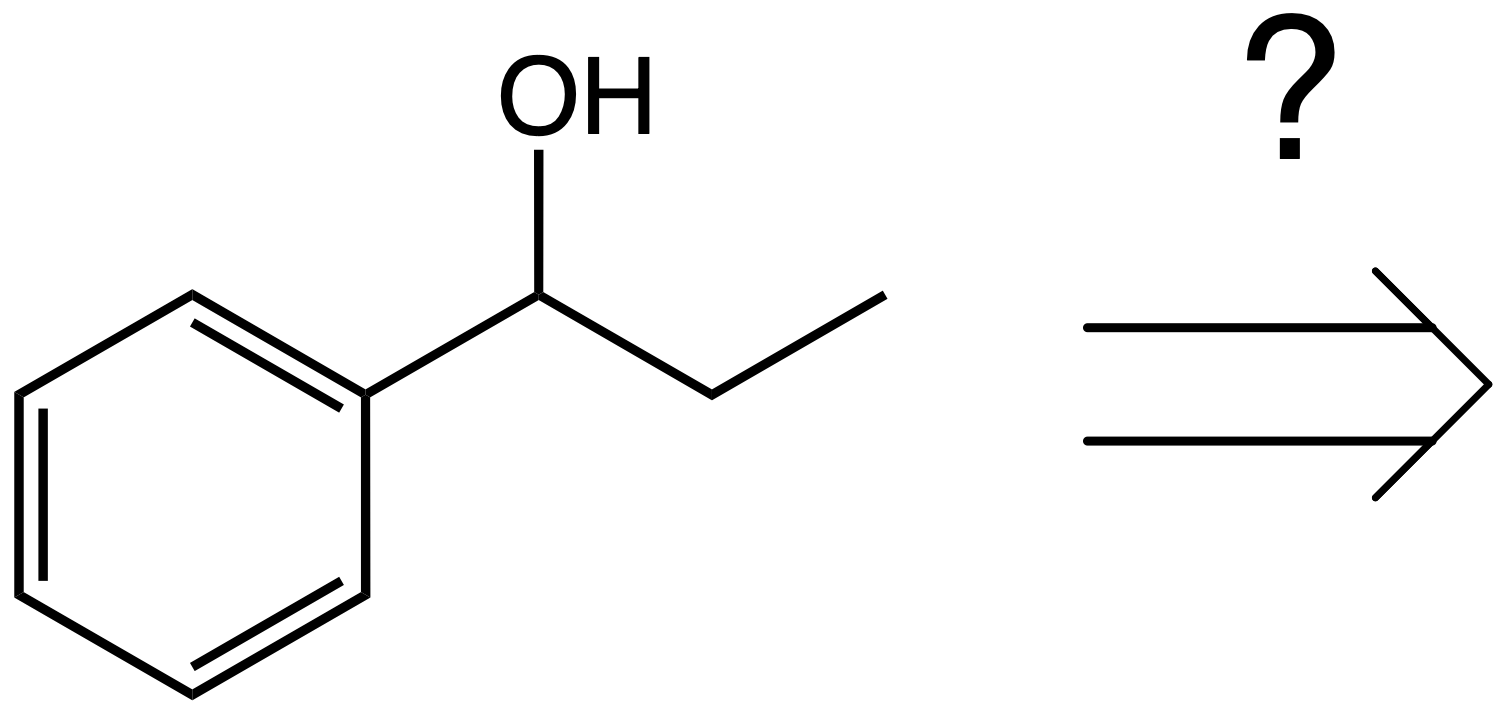
How do we decide this?
In the simplest approach, we would need to have a library of all possible transformations, and then we decide which transformations will more likely yield our desired product.
A list of possible transformations are all the name reactions you learn in your organic chemistry course: Grignard Reaction, Diels-Alder Cycloaddition, Aldolic condensation, etc. We then use our chemical knowledge to decide which of these can yield the product.
Clearly here, a Grignard reaction makes sense, while a Diels-Alder doesn’t.
Now, how can a model learn this process?
In this notebook, we will learn how to extract a library of transformations from a reaction dataset, and then train a model to do one-step retrosynthesis.
0. Relevant packages
RDChiral
RDChiral is a wrapper for RDKit’s functionalities for reaction handling, that improves stereochemistry handling. This package will allow us to extract reaction templates from a reaction dataset, which are a standard way of encoding transformation rules.
RDChiral then also lets us apply the reaction template to a target molecule, to discover the reactants that will afford the target molecule under the given transformation.
Download data
We will use a subset of the USPTO reaction dataset containing 50k reactions. This is commonly used to train and test new models for this task in the literature.
from utils import load_data, visualize_chemical_reaction
train_df, val_df, test_df = load_data()1. Reaction templates
Let’s take as an example the following coupling reaction.
rxn_example = train_df.iloc[5,0]
visualize_chemical_reaction(rxn_example)To extract the reaction template, use the extract_template function from utils.py
A reaction template describes a general transformation of some type. It describes what bonds form and break in a transformation, as well as the chemical environment of these bonds.
from utils import extract_template
tplt_example = extract_template(rxn_example)
# A reaction template looks like this
print(tplt_example)Now we can use this reaction template. Use the apply_template function from utils.py
If we use it on the same product, we should get the same reactants as above.
# Apply the extracted template to the product above.
from utils import apply_template, visualize_mols
prod_1 = rxn_example.split('>>')[1]
pred_reactants = apply_template(tplt_example, prod_1)
# This is the result of applying the template.
visualize_mols(pred_reactants[0])To see how reaction templates encode a general transformation, let’s now try to apply this template to a different product.
new_prod = 'CC1=CC(C2=CN=CN2C)=CC=C1'
# Let's try to break down this product, using the newly extracted reaction template.
visualize_mols(new_prod)new_reactants = apply_template(tplt_example, new_prod)
visualize_mols(new_reactants[0]) We have successfully encoded a Bromo Suzuki coupling!
As you can see, our reaction template can now be applied to any product, and the result of this operation are the reactants needed for this reaction to yield our product. You can learn more about these templates here.
What will happen if you try to apply the template to a product that makes no sense for this reaction type?
2. Building a library of reaction templates
Let’s now extract all possible reaction templates from our training data. With this, we can then train a model to select the best template for a given product!
This takes around 10 minutes.
import pandas as pd
from tqdm.notebook import tqdm
tqdm.pandas()
train_df['tmplt'] = train_df['reactants>reagents>production'].progress_apply(extract_template)
val_df['tmplt'] = val_df['reactants>reagents>production'].progress_apply(extract_template)
template_library = (pd.concat([train_df['tmplt'], val_df['tmplt']])
.value_counts()
.index
.values
)
print(f'We have {len(template_library)} different templates!')# Now assign a class number to each entry in the dataset
import numpy as np
train_df['rxn_class'] = train_df['tmplt'].progress_apply(lambda x: np.where(template_library==x)[0][0])
val_df['rxn_class'] = val_df['tmplt'].progress_apply(lambda x: np.where(template_library==x)[0][0])3. Training a ML model to do retrosynthesis.
We have over 11k templates, and for a given product we need to decide which one is the best. The best type of models for this type of task are classification models, and you’ve learnt a few of them at the beggining of the course!
In this case, instead of predicting whether a molecule is toxic (2 classes), we will predict what template to use to make the molecule (>11k classes).
As you may recall, we need to get a representation of our molecule that makes it easy for models to read it. Here, let’s use Morgan Fingerprints.
import mol_smi_to_count_fp from utils.py
from utils import mol_smi_to_count_fp
print('Getting FPs for training data')
train_fps = train_df['reactants>reagents>production'].apply(lambda x: x.split('>>')[1]).progress_apply(mol_smi_to_count_fp)
print('Getting FPs for validation data')
val_fps = val_df['reactants>reagents>production'].apply(lambda x: x.split('>>')[1]).progress_apply(mol_smi_to_count_fp)
print('Getting FPs for test data')
test_fps = test_df['reactants>reagents>production'].apply(lambda x: x.split('>>')[1]).progress_apply(mol_smi_to_count_fp)Now we can try to use ML to tackle the retrosynthesis challenge!
For this problem, classification models are quite appropriate, as we have a lot of classes and no idea how to assign them!
train_fps_dense = np.asarray(np.concatenate([v.todense() for v in train_fps.values]))
val_fps_dense = np.asarray(np.concatenate([v.todense() for v in val_fps.values]))# Let's define a very simple random forest classifier
from sklearn.ensemble import RandomForestClassifier
rf_clf = RandomForestClassifier(
n_estimators=300,
max_depth=15,
random_state=0,
n_jobs=20,
verbose=1
)
rf_clf.fit(train_fps_dense, train_df['rxn_class'])Testing the model
There are two ways to test our model: - Accuracy of template prediction - Accuracy of precursor prediction
As you will see, they can differ! Why?
# Let's see validation accuracy on template prediction
val_preds = rf_clf.predict(val_fps_dense)
val_acc = (val_preds == val_df['rxn_class']).mean()
print(f'Validation accuracy in template prediction: {val_acc*100:.3f}%')# Now, accuracy on precursor prediction
val_df['pred_tmplt_rfclf'] = template_library[val_preds]
# Apply the predicted template to the product
def get_precs_template(tmplt, prod):
pred = apply_template(
tmplt,
prod
)
if len(pred):
return pred[0]
else:
return 'Invalid template'
val_df['pred_prec'] = val_df.apply(
lambda x: get_precs_template(
x['pred_tmplt_rfclf'],
x['reactants>reagents>production'].split('>>')[1]
),
axis=1
)
#from utils import canonicalize_smiles
# Compare the resulting reactants
val_df['prec_true'] = val_df['reactants>reagents>production'].apply(lambda x: canonicalize_smiles(x.split('>>')[0]))
val_df['pred_prec'] = val_df['pred_prec'].apply(canonicalize_smiles)
prec_acc = (val_df['pred_prec'] == val_df['prec_true']).mean()
print(f'Precursor prediction accuracy is {prec_acc*100:.3f}%')# Pretty low, but why?
print(f'Template is invalid for {(val_df["pred_prec"]!="").sum()} products (out of {val_df.shape[0]}).')
# Let's see accuracy when template is valid
val_df_valid_preds = val_df[val_df['pred_prec']!='']
prec_acc_valid_preds = (val_df_valid_preds['pred_prec'] == val_df_valid_preds['prec_true']).mean()
print(f'Precursor prediction accuracy is {prec_acc_valid_preds*100:.3f}% (if we ignore invalid templates)')How to fix these issues?
Our current model predicts only one class. One way to mitigate this is to predict multiple classes, and return the prediction from the first valid template.
Train a better model.
The best models achieve over 55% accuracy on this dataset (see the leaderboard). There’s still a lot to improve on this simple model, try some of the things you’ve learnt during the course!
4. Let’s now apply our model to make some molecule of your interest
def predict_precursors(product):
fp = mol_smi_to_count_fp(product).todense()
logits = rf_clf.predict_proba(np.asarray(fp))
sorted_templts = np.argsort(logits)[0][::-1]
for i, t in enumerate(sorted_templts):
template = template_library[t]
try:
prec = get_precs_template(template, product)
except:
continue
if prec != 'Invalid template':
print(f'Using {i}-th template.\n\n')
print('To make this molecule')
visualize_mols(product)
print('The RF model predicts these reactants:')
visualize_mols(prec)
return prec
reacts = predict_precursors('CC1=CC(C2=CN=CN2C)=CC=C1')